胜利在望:即将完整的人类基因组
本文来自微信公众号:Nature Portfolio(ID:nature-portfolio),作者:Michael Eisenstein,题图来自:The Human Genome Project
由于基因和调控序列具有复杂多样的“地形”,人类基因组常常被比作为一幅地形图。但这幅图上有许多地方没有引人注目的美景,而是一望无际的沙漠公路。
以着丝粒为例,携带基因的两条染色体臂通过着丝粒相连。着丝粒由数千个几乎一样的α卫星序列组成——171bp的α卫星重复单位需要被正确排布以确保染色体稳定和细胞分裂。然而,在人类基因组草图发布20年后,着丝粒和其他有难度的DNA序列仍然是染色体图谱中难以填补的缺口。并且,直到几年前,一些研究人员还对填补它们感到绝望。
杜克大学的着丝粒研究员Beth Sullivan回忆起2014年与加州大学圣克鲁兹分校的基因组学研究员Karen Miga的一次谈话。“她告诉我,如果测序技术没有发生颠覆性改变,我们将在很长时间内停滞不前。” Sullivan说道。

纳米孔测序,例如GridION单元,能够不中断DNA而解码成千上万的碱基对。来源:牛津纳米孔科技。
但改变的确发生了:不间断读取长链DNA的测序技术横空出世。如今,Miga和从端粒到端粒联盟(Telomere to Telomere consortium,T2T)的同事即将完成一个长达20年的研究任务,这项工作是从基因组草图首次发布后就开始的。他们的目标是为每条染色体组装一个从端粒到端粒的基因组图谱,即从一个端粒(覆盖染色体末端的重复序列单元)延伸到另一个端粒。“这不仅仅是为了做而做,”Miga说到,“而是因为我觉得其中蕴含着重要的生物规律。” 但要找到它,基因组学界将需要对许多个这样的基因组进行测序,消除这些仍鲜为人知的基因组区域的变异。
进退两难
20年前的2月,人类基因组草图首次发表[1],这是一项里程碑式的成就,但它也有很多漏洞。“人类基因组计划”的科学家们从染色体DNA中获得了大量的短序列。这些短序列与相邻区域重叠,构成更大的连续序列——重叠群(contig)。理想情况下,每条染色体将呈现单个重叠群,但首次草图却包含了1246个这样的片段。
自此,基因组参考联盟(Genome Reference Consortium,GRC)的科学家们一直在完善组装,手动检查,并使用测序分析来识别有错误和信息缺口的片段。人类基因组图谱的最新版本于2013年发布,被称为GRCh38。从那时起,它就被反复修补。至今,它仍然缺少5%~10%的基因组,包括所有的着丝粒和其他困难区域,如编码核糖体RNA序列的大量基因。这些缺失的基因组藏于大量重复基因拷贝的长序列中。美国国家人类基因组研究所的生物信息学家、T2T联合主席Adam Phillippy说:“这在有待填补的缺口中占很大一部分。” 这些基因组还布满了几乎相同的难以比对的DNA序列,又称片段复制——这是古代染色体重排的产物。
这些难题继续阻碍着基因组的组装。这是因为到目前为止大多数测序都是通过短读长技术完成的,比如广泛使用的加州圣地亚哥生物技术公司Illumina商业化平台。Illumina测序仪可以获取非常精确的数据,但通常只有几百个碱基——因为太短而无法跨越长重复以及准确定位序列。“基因通常很容易组装,” 英国Wellcome Sanger研究所的计算生物学家、GRC成员Kerstin Howe说,“但基因间隔区里的其他序列或有很多重复基因的序列却很难着手。”
填补缺口
两种长读长测序技术正在填补这些缺口。加州生物技术公司太平洋生物科学(Pacific Biosciences,以下简称PacBio)使用一种成像系统来直接读取数十万甚至数百万条平行DNA链,每条链包含数千个碱基。另一种技术是由英国公司牛津纳米孔技术(Oxford Nanopore Technologies)实现商业化,它将DNA链穿过微小的蛋白孔或纳米孔,测量核苷酸穿过孔道时电流的细微变化,进而读取数万至数十万个碱基。
当这两项技术首次推出时(PacBio于2010年、牛津纳米孔于2014年推出),它们比Illumina更容易出错,Illumina单次读长的准确性超过99%。“我们在说,早期PacBio 读长错误率为15-20%,”Phillippy说。而第一代nanopore测序仪会在超过30%的碱基中产生误差。
但这些技术在稳步改善,其读长也随之提高。“在过去三四年间,我们可以读取超过100千碱基(kb)的序列长度,” Phillippy说,“也就在那时,Karen和我成立了T2T联盟。”
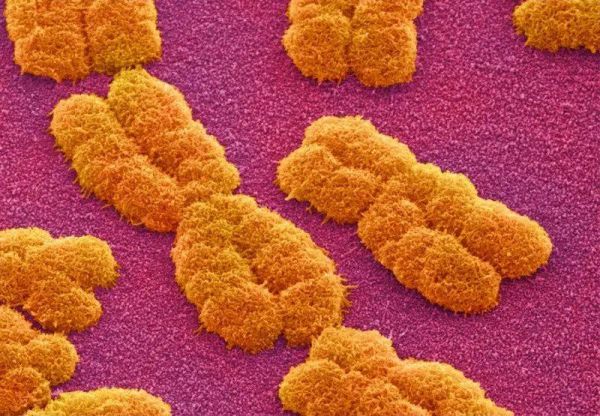
人类染色体的扫描电子显微镜成像。来源:Power and Syred/SPL
T2T联盟成立于2019年初,旨在为每个人类染色体提供高质量的从端粒到端粒的组装。来自世界各地的100多名测序和基因组学专家已经与联盟签约,其中许多专家积极展示了长读长测序分析的优势。
2018年发表的两篇论文突出了他们的工作。在第一篇研究中[2],英国诺丁汉大学的计算生物学家Matthew Loose和同事描述了第一个完全根据Oxford Nanopore数据组装的人类基因组。过去,长读长组装是使用Illumina数据来纠正容易出错的nanopore结果。但是,现在Loose和同事仅仅使用Nanopore数据就覆盖了大约90%的GRCh38,准确率高达99.8%,同时也填补了参考基因组中的十几个主要缺口。
在第二项研究中[3],Miga和她的团队重新组装了人类Y染色体的着丝粒,这是基因组中最小的染色体。他们在该区域合成了大量的长读长序列,获得了高质量的共有序列,其中的随机错误很容易被识别和消除。“实际上我们可以横跨整个着丝粒,”Miga说,“但在那时工作仍然是手动完成的——就是看着模式然后把它们连接在一起。”
首战告捷
这些成功表明T2T的目标是可以实现的。为了简化工作,T2T联盟专注于CHM13,这是一种肿瘤衍生细胞系,其基因组包含两套相同的染色体。这就避免了二倍体基因组的复杂性,因为二倍体基因组具有来自父母双方的不同染色体拷贝。
在2020年底,T2T科学家公布了两个染色体的完整组装,即X染色体[4]和8号染色体 (预印本)[5]。研究人员使用牛津纳米孔技术,对两条染色体的片段进行测序,这两条染色体的长度通常超过7万个碱基,其中一条读长甚至超过100万个碱基。Phillippy说:“有了这些,我们就能够获得从端粒到端粒的染色体的主干展示,但精确度较低。” 然后,他们用Illumina和PacBio的读本作为补充,以优化组装。
华盛顿大学基因组学家Evan Eichler实验室的博士后Glennis Logsdon是8号染色体研究工作的第一作者,他说不同的测序技术都有其不足。例如,T2T科学家们发现,PacBio会在G和A碱基高度富集的基因组区域出错,而nanopore有时会在相同核苷酸的长重复序列中出错。Logsdon说:“如果一个数据库有某种缺陷而另一个数据库没有,那么它们最终会因此而很好地互补。”
完成和检查基因组组装需要专用的软件,这套工具由Phillippy和加州大学圣地亚哥分校计算生物学家Pavel Pevzner等研究人员共同开发。研究小组采取谨慎的方法,“只有两个长度超过7000个碱基的序列,基本上100%相同,我们才会把它们粘合在一起,” Phillippy说,“因为一旦你在组装中引入一个错误,就很难修复它。” 他说,通过这么谨慎的操作,有可能在核苷酸水平上产生99.99%的准确组装。
对X染色体的初步研究[4]也得益于之前对该染色体着丝粒的了解,此着丝粒的结构已经被充分研究。“我们根据测序信息来组装α卫星序列,使用多种分子技术确保其大小合适,” Sullivan说,“总的来说,我对第一次研究中的验证工作量印象深刻。”
研究人员还利用了作图技术,比如加州生物科技公司Bionano Genomics开发的一种技术,使得测量一条染色体上不同DNA序列之间的距离成为可能。
接近完成
虽然很成功,但是T2T方法对8号染色体和X染色体的测序过程费力且艰苦。在这段时间内,一个重要进展给团队的努力打了一剂强心针。PacBio仪器支持环形一致测序(CCS),在此过程中单个DNA链被转换成可以反复读取的闭环。通过比较这些重复序列,研究员可以消除随机错误,获取高度准确的结果。
早期版本的CCS最多容纳几千个碱基,在基因组组装中用途有限。但2019年,PacBio改进了这一过程[6],后续的高保真技术如今产生超过2万个碱基的一致序列,准确率超过99%。Pevzner说:“我们现在组装出的一些着丝粒完全来自高保真序列,而不需要额外的数据。” 不过他随后补充道,另外还需要精确校准的算法来处理这些数据。
Pevzner将着丝粒重组比作拼接看似清晰的蓝天拼图,在这个拼图中,最初所有的碎片看起来都差不多。他说,“里面有少少的、几乎看不见的云朵,能用来分辨拼图的不同部分。” 找出这些云揭开了拼图的拼法,改进后的方法对着丝粒也有类似作用,即敏锐检测到细微的序列差异,为组装算法提供标记。
这种方法与更长的nanopore测序结合后显著加速了T2T进展——Logsdon报告称十万个碱基的延伸现在已经十分普遍。“我们花了一年甚至更多的时间来完成X染色体和8号染色体的研究,”Phillippy说,“但后来我们基本上在两个月内就完成了剩余所有染色体的工作。” 如今,终点近在眼前。“我们搞定了所有着丝粒序列,除了9号染色体上的一个,”Miga说,这个着丝粒太大了,跨越2700万个碱基,验证特别难。该团队还在最终确认高度复制核糖体RNA基因。但T2T已经在GitHub上分享了数据,Miga预计CHM13细胞系的完整基因组将于今年发布。
这些数据加深了人们对基因的理解。Logsdon和其他人一直在使用nanopore测序来发现能够影响染色体功能的DNA化学修饰模式。她说,“大多数着丝粒都发生了甲基化,但似乎在所有着丝粒中都存在这种甲基化标记。” 甲基化可能标记了动粒的位置,这是一个重要的着丝点结构,负责细胞分裂期间DNA的均匀分配。Logsdon希望利用这些发现,设计用于合成染色体的最小着丝粒。
T2T的方法也使得庞大复杂的基因测序工作相对缩短,如编码免疫系统T细胞表面抗体和受体可变区的基因。“它们是高度重复的,而且组装起来非常困难,” Pevzner说,“目前为止,我们只有两个可变区的参考序列。” 获取和表征这些具有挑战性的基因组片段将引导我们理解感染和疫苗的免疫应答机制。
顺利完成
尽管组装那么难,但如果没有与不同个体的其他基因组进行比较,单一的从端粒到端粒的基因组提供给研究员的价值有限。为提高效用,在2020年末,T2T开始与人类泛基因组参考联盟(HPRC)展开紧密合作。HPRC于2019年成立,旨在用一个能更好记录人类多样性范围的参考基因组取代GRCh38,该参考基因组基于至少350名个体的全基因组数据。德国马克斯·普朗克信息学研究所的计算生物学家Tobias Marschall参与了这项研究,他说:“基因组医学越是成为常规,你就越想消除某人从祖先那儿继承来的任何偏差。”
东京大学计算生物学家森下真一(Shinichi Morishita)实验室的研究助理铃木裕太(Yuta Suzuki)利用PacBio测序研究了来自日本和世界其他地区的36个人的着丝粒[7]。“就在日本人群中,我们发现几乎每一个我们调查过的样本都有不同的着丝粒,” 铃木说,“所以只有一个参考是不够的,甚至每个群体有一个参考都不够。”
森下计划分析另外几百个人类着丝粒,并指出几十种疾病相关的基因变异已经被定位到这些区域。他说:“这表明丝粒重复序列中月问题,我们初步想法是它们的稳定性可能由于结构突变而被破坏了。” 在Phillippy看来,一旦核糖体RNA基因可以常规处理,就能更好地理解与细胞蛋白质合成机制相关的疾病。
但首先,研究人员必须弄清楚如何将T2T过程应用于二倍体基因组。要确定哪条序列位于哪条染色体上,科学家需要发现足够独特的遗传标记,进而为每条DNA链组装独特的重叠群,但这是一项艰巨的任务,尤其是着丝粒这样的超重复区域。在他们的8号染色体预印本中,Logsdon和Eichler等人描述了在黑猩猩和人类中重组二倍体着丝粒区域的可行性,但前提是这两条染色体的基因有高度独特性。Morishita说:“对于二倍体基因组,我们需要更精确的或更长的读长来横跨整个着丝粒区域。”
目前,大多数临床-基因组学研究集中于已知的基因,这是一种快速经济的基因组分析方法。然而,探索这一新领域的先驱们预计,尽管综合分析可能花费更多,但它最终将成为医学和基因组学研究中的一个标准,尤其是当研究人员开始例行地探索这些过去未比对区域基因变异的临床影响。“如果我的孩子生病了,而我知道我可以通过长读长测序得到100%的基因组的话,我会愿意支付增加的费用的。”Miga说。
参考文献:
1. International Human Genome Sequencing Consortium. Nature 409, 860–921 (2001).
2. Jain, M. et al. Nature Biotechnol. 36, 338–345 (2018).
3. Jain, M. et al. Nature Biotechnol. 36, 321–323 (2018).
4. Miga, K. H. et al. Nature 585, 79–84 (2020).
5. Logsdon, G. A. et al. Preprint at bioRxiv https://doi.org/10.1101/2020.09.08.285395 (2020).
6. Wenger, A. M. et al. Nature Biotechnol. 37, 1155–1162 (2019).
7. Suzuki, Y., Myers, E. W. & Morishita, S. Sci. Adv. 6, eabd9230 (2020).
原文以Closing in on a complete human genome标题发表在2021年2月22日的《自然》的技术特写版块上
本文来自微信公众号:Nature Portfolio(ID:nature-portfolio),作者:Michael Eisenstein
相关推荐
胜利在望:即将完整的人类基因组
挑战造物主的“科学极客”,差点就破解了人类基因组
生物技术和生物经济:大放光彩的十年即将到来
基因测序:比肩移动的下一个“平台技术”(二)
人类正向“造物主”靠拢:基于CAD的全新基因编辑软件,让设计新生命像画画一样简单
基因测序:比肩移动的下一个“平台技术”(一)
利用新一代TILs疗法治疗实体瘤,「劲风生物」完成一千万美元Pre-A轮融资
妈妈如何影响子女身高、寿命?剑桥科学家找到新证据,或有望帮助后代长高、长寿
重复的基因:你我都是复读机
这种基因中的“废物”,蕴含着抗击癌症的巨大潜能
网址: 胜利在望:即将完整的人类基因组 http://www.xishuta.com/newsview43875.html
推荐科技快讯






- 1问界商标转让释放信号:赛力斯 95254
- 2人类唯一的出路:变成人工智能 21423
- 3报告:抖音海外版下载量突破1 21393
- 4移动办公如何高效?谷歌研究了 20566
- 5人类唯一的出路: 变成人工智 20563
- 62023年起,银行存取款迎来 10359
- 7五一来了,大数据杀熟又想来, 8806
- 8网传比亚迪一员工泄露华为机密 8543
- 9滴滴出行被投诉价格操纵,网约 8429
- 10顶风作案?金山WPS被指套娃 7247



