这些天价救命药将退出中国,发生了什么?
本文来自微信公众号:CC情报局 (ID:cancer-weekly),作者:虞梦奇,原文标题:《这些患者的天价救命药将退出中国,什么原因造成了这一困局?》,题图来自:《黑客帝国:矩阵重启》
核心提要:
1. 近期传出消息,用于治疗黏多糖贮积症IVA型(MPSIVA)的特效药“唯铭赞”将退出中国市场。MPSIVA是一种典型的罕见病,症状包括生长迟缓、脖子异常短等。国内已知的该病患者在100人左右,虽然其中使用“唯铭赞”的患者只有十几人,但该药退市意味着这些患者继续用药将面临困难。
2. 药品生产商百傲万里制药给出的退市理由是,“复杂的市场准入规则使得药物的供应不可持续”。“唯铭赞”于2019年5月在中国获得上市批准,目前仍未纳入国家医保目录,其在中国的定价为7500元/支(5mg),一个25斤的孩子,一年用药的总价在200万元左右。
3. 罕见病仅在极少数人身上发生,治疗罕见病的药物叫作孤儿药,其商业模式采用高价策略,原因是开发罕见病药物的企业必须掌控高定价,从而保证财务可持续。但由于孤儿药上市速度加快,高昂药价给医保带来的压力越来越大。
4. 短期内将所有罕见病高值药纳入基本医保并不现实,相关研究人员认为,“多方共付”模式可以保障患者用药可及,该模式在患者、制药厂商和第三方财政支持机构的三方架构内,通过扩展第三方资源来解决成本问题。
罕见病神药在中国获批上市不到四年,为何要退市?
一种治疗黏多糖贮积(MPS)的超级罕见病药物“唯铭赞”(Vimizim,依洛硫酸酯酶α),是目前世界上唯一获批用于治疗黏多糖贮积症IVA型(MPSIVA)的特效药。2019年6月在中国批准上市,该药是国内首个黏多糖贮积症治疗药物,被列入我国第一批《临床急需境外新药名单》。截至目前,在MPS多个亚型中,共有3款特效药在国内获批,针对I、II、IV型,年均费用均过百万。
但令人震惊的是,近期关于这家药企即将退出中国,并不再销售“唯铭赞”的消息,在病人中流传,并引发恐慌。6月8日,媒体就此向“唯铭赞”的药品生产商百傲万里制药(BioMarin Pharmaceutical Inc)进行了查证,得到了证实:“百傲万里已决定不再为‘唯铭赞’在中国的进口药品注册证(IDL)续期,目前该注册证将于2024年5月到期。”这意味着,这款药将从中国退市,可能导致国内使用该药治疗罕见病黏多糖贮积症IVA型(MPS IVA)的患者继续用药遇到难题。
这一消息令人揪心,尽管全国此类疾病患者数量并不是很多,可这毕竟就意味着少数家庭的苦难。但是,这一消息也并不令人意外。坦白地说,这是世界上所有罕见病患者随时都可能遇到的难题。
治疗这类疾病的药物一般被称为孤儿药,其商业模式决定它必然昂贵,为了分摊成本,必然在患者、制药厂商和第三方财政支持机构之间形成博弈。其中患者肯定是最弱势的,而厂商和第三方则肯定有不同的利益考量。
比如UniQure公司研发的用于治疗脂蛋白脂酶缺乏遗传病的药物Glybera,是2012年欧洲药品管理局批准的首个基因疗法,但是由于其每剂130万美元的极高定价,导致了各国都面临巨大医保报销压力,没有一家政府愿意为其买单,该药物最终在2017年起就未更新过上市授权。
事实上,在讨论这个复杂的问题前,人们需要先理解两个基本概念:
1. 罕见病。又称罕见疾病,按字义理解,就是指仅在极少数人身上发生的稀罕病症,也被称为孤儿病。但具体如何定义极少数,世界上并无统一标准。美国国家卫生院的标准是,全美总病例数少于20万例,以此计算,全球大概有7000种罕见病,影响2500万到3000万美国人。
而上世纪80年代开发出治疗罕见病商业模式的美国制药公司健赞(Genzyme)的定义是,患病人口少于1万例。在我国,根据《中国罕见病定义研究报告2021》,罕见病最新定义是指新生儿发病率小于万分之一、患病率小于万分之一、患病人数小于14万的疾病,官方估计中国罕见病患者有2000万人。
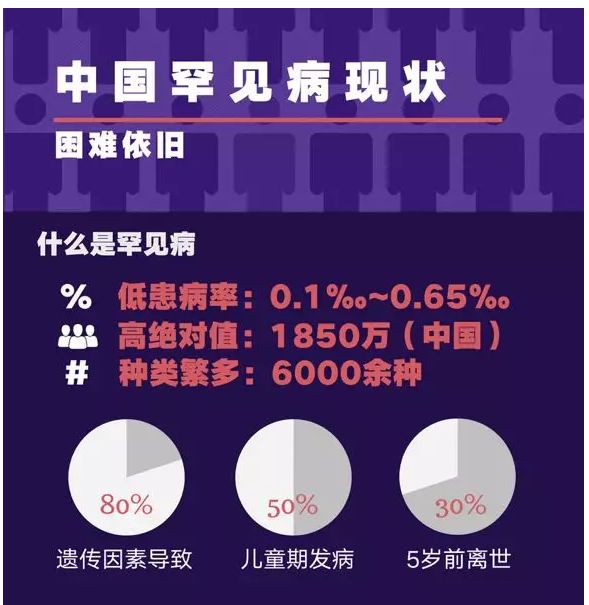
图/《中国罕见病定义研究报告2021》
2. 孤儿药。又称为罕见药,用于预防、治疗、诊断罕见病的药物、诊断试剂和疫苗。由于罕见病患病人群少、市场需求少、研发成本高,以前很少有制药企业关注和研发孤儿药,但是后来出现了孤儿药商业模式。
这种模式源于三个要素:一是罕见病总体上缺乏治疗方案,目前世界上大约只有500种罕见病可以治,故奇货可居;二是罕见病经常源于遗传异常导致酶变异体缺失或功能失常,治疗方案通常是价值几十万美金的酶替代疗法,单价很高;三是大部分的罕见病都是遗传病,特效药将伴随患者终生,治疗周期往往比癌症要长得多,用药总价值很高。
破解罕见病的治疗困境可能并不容易
黏多糖贮积症IVA型(MPS IVA)便是一种典型的罕见病。它是已知的40多种不同的溶酶体贮积病的一种,是Morquio综合征,即黏多糖贮积症IV型或MPS IV的一种形式。
Morquio综合征分A和B两种形式,分别是由于N-乙酰半乳糖胺-6-硫酸酯酶和β-半乳糖苷酶缺乏,导致A型角质素和硫酸软骨素问题,以及B型硫酸角质素问题。任何一种酶的缺乏都会导致体内粘多糖的积累、骨骼发育异常和其他症状。
在大多数情况下,患有Morquio综合征的人智力正常,B类临床特征通常比A类更少且更轻微。其症状可能包括生长迟缓、突出的下脸、异常短的脖子、异常靠拢的膝盖(膝外翻或膝外翻)、扁平足、异常的侧向和前后或左右弯曲脊柱(脊柱侧凸)、长骨生长末端异常发育(骨骺)和/或突出的胸骨(鸡胸)。 在某些情况下,还会出现听力损失、腿部无力和/或其他异常。
治疗黏多糖贮积症的特效孤儿药出现得比较早。2005年,美国食品和药物监督管理局批准半乳硫酶 (Naglazyme)用于治疗MPS VI,也称为 Maroteaux-Lamy综合征,这款药就是百傲万里的产品。2003年,FDA 批准 laronidase(Aldurazyme)为MPS I的治疗药物,用于患有Hurler和 Hurler-Scheie型MPS I的患者以及表现出中度至重度症状的Scheie型患者,这款药还是百傲万里造的,由Sanofi-Genzyme分销。
此后,FDA还批准了用于MPS II的艾杜硫酶(Elaprase,2006年7月)、用于MPS VI的半胱氨酸酶(Naglazyme,2005年5月)和用于MPS IVA的elosufase alfa(Vimizin,即“唯铭赞”,2014年2月)。Elaprase由Shire Pharmaceuticals生产,Naglazyme和Vimizin均由百傲万里生产。
据国内媒体报道的数据,黏多糖贮积症IVA型在国内已知的患者大约100人,使用“唯铭赞”的患者只有十几人。“唯铭赞”2018年在中国大陆申请上市,借着中国政府对罕见病药物开辟的绿色通道,2019年5月就获得了国家药品监督管理局的上市批准,被列入我国第一批《临床急需境外新药名单》。作为高值药物,“唯铭赞”曾出现在2021年的医保谈判预选名单中,但最终谈判失败,2022年未参与医保谈判,因此目前该药仍未纳入国家医保目录,只纳入了部分地方医保及惠民保。笔者查到的可覆盖城市包括杭州、济南、德阳、东营、肇庆、烟台和无锡等。
“唯铭赞”非常贵,在中国的定价为7500元一支(5mg),需要依据患者体重决定用药剂量,一个25斤的孩子,一年用药的总价在200万元左右。2023年2月27日,百傲万里制药发布2022年财报,全年营收20.96亿美元,“唯铭赞”去年全球营收6.64亿美元,同比增长7%,但在中国大陆市场营收空间占比很小。

百傲万里制药给出的“唯铭赞”退出中国市场的理由是:“复杂的市场准入规则使得药物的供应不可持续,特别是在罕见病治疗方面。尽管我们在过去几年中尽了最大努力,仍然没有促使药品进入医保报销体系,我们决定不再续签该产品的进口药品注册证。”
作为企业,不论是依据市场状况,还是依据相关成本考量,进入和退出市场的选择都是正常现象。特别是孤儿药商业模式,其构建基础是必须有人买单。1994年,Genzyme推出治疗戈谢病的孤儿药Cerezyme(伊米苷酶)时,在美国对普通患者的定价约为每年20万美元。这在当时是一个很高的价格。 历史数据显示,Cerezyme上市价格比同期新型慢性药物的平均成本(825美元/年)高出2400% 以上。
实际上,Cerezyme恰恰发明了高价罕见病商业模式。它的理念是,如果该行业要始终如一地为罕见病开发药物,就必须掌控高定价,使其成为财务上可持续的业务。这种定价策略假设,由于Cerezyme和其他孤儿药影响的患者很少,医疗保健系统可以相对轻松地消化高价,在此过程中,服务严重不足的人群可以获得急需的护理。但是,随着新型生物技术对罕见病的认识深入,孤儿药模式扩展,上市速度急速加快,医保为此所能承受的压力也就越来越大。
美国调查公司数据显示,从2017年到2021年,美国新上市数十种孤儿药的中位数批发价格是21万8872美元,而数百种非孤儿药新药的批发价格中位数是1万2789美元,一共只有7种非孤儿药价格达到了孤儿药的中位数水平。当前,美国正呈现孤儿药支出爆炸的趋势,全美药品消费市场规模2022年为5180亿美元,孤儿药2022年已达到1730亿美元,预计2026年达到2740亿美元。
Cerezyme这些成功的企业也在扩展思路,它称为“高接触”服务(high-touch)的战略,就是向国际患者社区密切合作,来了解某一种罕见病的流行病学数据以及治疗结果,然后为患者做登记,提供财政和组织支持,这样即可以既完成患者识别,也树立了药物和厂商在医生和患者群体中的广泛认可。
为何无法全部纳入医保?罕见病人如何获得救命药?
拓展市场是另一个拓展方向。在“唯铭赞”考虑退出中国大陆市场的时候,其它孤儿药在不断进入。
据病痛挑战基金会统计,2019年至2021年有32个罕见病药物已在中国大陆获批上市,已有12个罕见病药物进入医保,这12种药物从上市到进入医保的平均时间约为14个月。当然,考虑到国家基本医保基金的承受能力、社会公平性的要求、企业的中国市场定价策略等因素,短期内将所有罕见病高值药纳入基本医保并不现实,更不要说应对类似美国市场现在的孤儿药支付爆炸的情形,这让那种建议在医保总盘子内保留一定比例专门用于罕见病的想法变得没有可持续性。相关行业研究人员认为,部分罕见病药物没能得到基本医保的保障,则需要运用其他途径,通过“多方共付”模式保障患者用药可及。
这其实就是在患者、制药厂商和第三方财政支持机构三方架构内通过扩展第三方资源来解决成本问题的思路,这也是许多国家的普遍思路。在第三方领域,我国的情况相对比较简单,主要是国家全民医保、地方医保和商业保险,在美国情况则更加复杂,因为制药产业、保险业乃至连锁药店在中间具有非常大的话语权,相互之间还要展开谈判,而且相互之间存在不透明。
举个例子,2022年,管理着美国80%的所有处方药的三大采购商,从其标准商业保险处方药集中排除了1150多种药物,这意味着自2014年以来在排除商业保险外的药物增加了近1000%,其中包括能够以较低成本为患者提供所需治疗的药物。
总体来说,美国各类医保对孤儿药的覆盖率还是比较高的,可以达到七成,具体比率,美国政府公共卫生机构专门拨款展开的调查也搞不清楚,因为上市孤儿药越来越多,而保险套餐成千上万,不同罕见病和孤儿药又在名单中进进出出。但罕见病患者普遍感觉现在的保险所提供的药物和其它医疗服务变得更少和更贵。患有庞贝病的作家Keyana Sullivan最近写道:
“你想知道这一切的疯狂之处吗?当我们谈论这样的事情时,每个人的反应总是‘他们不能那样做’。好吧,他们当然可以这样做,因为一切都与金钱有关。医学界再神奇,一半是未来科技,一半是赚钱。当你依赖别人而不给人家回报时,你最终别无选择,只能接受人家给你的东西。这就是医疗保险的感觉。”
所以,更复杂的针对罕见病和孤儿药的结构性创新正在被探讨。它主要从四种模式入手:非营利性制药公司模式、运营虚拟公司将投资者与生物制药公司联系起来的罕见病基金会、由在罕见疾病领域拥有既得利益的盈利公司共同分担模式,以及致力于推进药物发现的全球私募股权基金。这些制度性创新也可以为我国所借鉴,它的思路实际上是从研发端降低和分摊罕见病治疗的成本困境。
本文来自微信公众号:CC情报局 (ID:cancer-weekly),作者:虞梦奇
相关推荐
天价“救命药”跌到“地板价”,中国创新药开打翻身仗
疫情不是在线教育的救命药
垄断心梗“救命药”,百亿药企被罚没2.85亿
竟将海洛因合法化,美国发生了什么?
50余款抗癌药物获FDA审批,“救命药”何时能不再遥不可及?| 2020盘点
处境急转直下,特斯拉今年到底发生了什么?
新股上市接二连三破发,发生了什么?
从“收购亚马逊中国”到被阿里收购,网易考拉发生了 什么?
共享充电宝集体涨价,背后到底发生了什么?
18位股东集体退出,还不上市的叮当快药能否找到突围的快药方
网址: 这些天价救命药将退出中国,发生了什么? http://www.xishuta.com/newsview78631.html
推荐科技快讯




- 1问界商标转让释放信号:赛力斯 95088
- 2人类唯一的出路:变成人工智能 20304
- 3报告:抖音海外版下载量突破1 20127
- 4移动办公如何高效?谷歌研究了 19524
- 5人类唯一的出路: 变成人工智 19425
- 62023年起,银行存取款迎来 10245
- 7网传比亚迪一员工泄露华为机密 8365
- 8五一来了,大数据杀熟又想来, 7851
- 9滴滴出行被投诉价格操纵,网约 7477
- 10顶风作案?金山WPS被指套娃 7167



