14:1止步FDA,信达PD-1出海受挫释放了什么信号?
2月10日,信达生物和礼来制药合作的国产PD-1肿瘤药Sintilimab(信迪利单抗)首次直面FDA肿瘤药物咨询委员会(ODAC),最终,专家委员会以14:1的投票结果反对申请,宣布中国首位闯关FDA的PD-1"选手"需要暂时止步。
争议主要在于2点:
试验人群/数据:FDA专家认为使用单一国家数据不足以证明对美国患者的有效性;
试验终点:此前FDA批准肺癌一线疗法用的都是总生存期(OS),而该试验用的是无进展生存期(Progression-Free Survival,PFS)。
为此,委员会投票建议在批准之前,必须进行另一项临床试验(OS+头对头),以证明该药对美国患者有效。
这意味着,该药物也许还需要为期7年的试验周期和高额的费用支出,才有可能在美国上市。
一叶知秋,FDA正在收紧对PD-1的审核,而信达生物不幸第一个“撞到了枪口”,那些同样手持PD-1的后来者进退何如?国产创新药出海是否会就此止步?
“FDA:为中国药企立规矩”
此次申请被拒的结果并不突然。
去年12月,FDA癌症药 物负责人Richard Pazdur博士 在接受媒体采访时就曾表示:利用单一国家临床数据的上市申请存在问题,这与美国为增加临床试验患者多样性所做的努力背道而驰。

▲Richard Pazdur博士
该言论被认为是FDA审批收紧的重要信号。
本月4日,Pazdur再次发文,再次重申关于单一国家临床试验数据PD-1在美国上市条件。
这距离信达生物的过会申请仅剩不到一周时间。
在2月10日的会议上, FDA和ODAC专家火力全开,指出了Sintilimab的五处“硬伤”:

▲信达ODAC会议纪要,来自:海通医药(只供参考)
值得一提的是,在整场讨论中,未见到对Sintilimab有效性和安全性的“实锤”挑战,说明药物实力经受住了考验。
为争取审批,礼来承诺,如果信迪利单抗能上市,他们将在美国提供40%左右的折扣(默沙东的PD-1 Keytruda年售价为15万美元),同样有一位专家,也是全场唯一一位投赞成票的专家指出, 便宜、有效的治疗方法应该被鼓励进入美国。
然而,FDA态度非常“头铁”,明确回应, 价格因素并不是批准与否的考量标准。

价格优势是中国药企在美市场的“王牌”之一,FDA一句“不考虑”,直接腰斩众人的希望。
此外,在会议陈述环节,FDA 也感受到了来自大洋彼岸的“内卷”压力:越来越多的肿瘤开发项目完全或主要基于来自中国的临床试验和数据,其中 至少有 25 个 申请计划提交或目前正在 FDA 审查。
在这场论证会上,FDA决定顶住压力,不为此类审批开放先例。
以上各类陈述,于中国药企的针对性不言自明,这让人很难不联想到这场审评会议 或是FDA为单一或中国临床研究数据为主的新药上市申请来“立规矩” 。
出海新门槛——全球多中心临床试验
其实,长久以来,中国NMPA和美国FDA在新药审批中,对境外新药临床数据都是默认许可的。
根据ICH(国际人用药品注册技术协调会)E5 R1规定,允许将境外临床数据外推至新地区人群。
外企正在积极地将中国纳入全球临床早期阶段,这是在华MNC加速全球新药同步上市的策略之一,比如:
2009年,上海中心加入罗氏全球药品开发中心,位列全球TOP5。自此,罗氏所有创新药的研发都会同时放在中国,并已基本做到与全球三期临床同步。
2021年,阿斯利康位于上海的全球研发中国中心正式启用,并作为全球研发网络的重要成员深度参与其全球新药同步研发。
2022年,辉瑞将把全球超八成关键临床研究落地中国。
武田亚洲开发中心目前全球40个临床阶段在研产品中已有25个具有中国计划,剩下的产品后续也会带到中国。
而这对实力较薄弱中国药企来说,却是“沉重的负担”。
FDA的门槛在于,接受来自境外的临床试验数据,但要求支持性的临床里面有美国人种的代表性。
FDA会对临床数据中的亚组进行趋势分析,如果某个亚组与整体趋势一致,至少有30%入组病例来自欧洲、美洲的白人,则可认为这个临床试验的数据具有美国人种代表性。
简单来说,就是临床试验关键不是在哪里做,而是入组病例有多少白人。信达生物信迪利单抗海外人种入组比例只有3%(白种人入组更低)。
因此,全球多中心尤其是多人种临床试验或将成为新药获批必选项,这将成为中国药企出海必须面临的考验之一。
当然,也有例外的情况,比如部分罕见病,如在美国招募困难,FDA审查人员表示 "监管也可更灵活"。
创新药出海元年“夭折”?
2022年,本应是创新药国际化元年。
首创证劵数据显示,2021年,有6款国产药物通过FDA申请受理,预计审批时间集中在2022年,其中有4款PD-1产品。
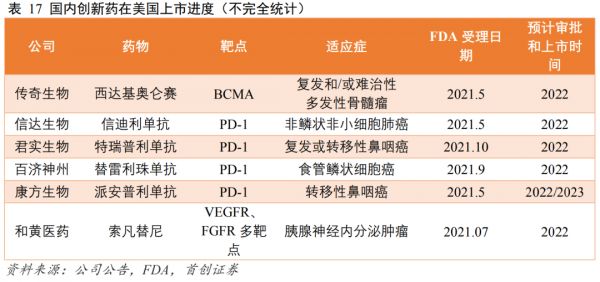
其中君实生物是跟信达生物同期,最早申报的产品之一:
去年3月,君实生物向FDA滚动提交了PD-1用于治疗复发或转移性鼻咽癌(npc)的上市申请,目前美国尚无PD-1单抗获批用于鼻咽癌的治疗,君实是这个适应症的潜在First in class;
10月,特瑞普利单抗联合吉西他滨╱顺铂作为晚期复发或转移性鼻咽癌患者的一线治疗和单药用于复发或转移性鼻咽癌含铂治疗后的二线及以上治疗的两项适应症的生物制品许可申请(BLA)获得FDA受理,这也是首个向FDA提交上市申请的国产抗PD-1单抗。
根据君实披露的信息,该申请的临床试验包含国际多中心临床,但不知是否是以中国为主的亚洲患者数据。

此外,恒瑞医药、百济神州、康方生物PD-1多中心临床试验的白种人病例入组率都不到30%。
因此,信达不太可能是唯一受这一裁决影响的公司,其他几家公司面临同样的挑战。
实际上,中国创新药完全基于美国临床数据获FDA批准上市的,目前还寥寥无几。
从已经通过审批的药品来看,2019年底,百济神州的泽布替尼和石药集团的玄宁均是主要依据中国的临床数据。
而对于全球大三期,国内药企不是不想做,而是很难做得起。
海外临床费用巨大,特别是做头对头试验。据百济神州全球研发负责人汪来介绍,2018年底,百济神州启动泽布替尼头对头伊布替尼的ALPINE研究,伊布替尼当时在美国对慢性淋巴细胞白血病的治疗费用大约在12万美元/年,且入组的患者中,300多名是使用伊布替尼的患者,许多患者需要用药几年,买药的费用加上临床本身的费用十分巨大。
汪来曾说,“好多人说我们烧钱,但对于药企而言,有时是无法回避的,因为你想证明药物是国际领先的话,就不得不投入。”
国际化是方向,实力与耐心的考验已经到来。
万春医药普那布林、信达PD-1已经受挫,百济神州、君实生物、康方生物、和黄医药索凡替尼、传奇生物CAR-T西达基奥仑赛等大考也不远了。
本文来自微信公众号 “MedTrend医趋势”(ID:Trendhc),作者:更多精彩资讯,36氪经授权发布。
相关推荐
14:1止步FDA,信达PD-1出海受挫释放了什么信号?
PD-1年费用进入1万元区间,国产创新药内卷和消失的千亿市场
国产PD-1抗癌药的好日子到头了?
“肿瘤神药”PD-1红海中,恒瑞、百济、信达、君实四小龙谁拔头筹?
22天粉丝突破350W,硬核老师B站爆红背后释放了哪些信号?
小米再次人事调整,这背后释放了什么信号?
PD-1“鸳鸯谱”大全
阿里欲百亿入股申通快递,电商巨头释放了什么信号
气象超脑,释放了一只混沌蝴蝶
“决战双十一”,120万抗癌神药或无缘医保谈判,国产PD-1药企股价飙升
网址: 14:1止步FDA,信达PD-1出海受挫释放了什么信号? http://www.xishuta.com/zhidaoview23845.html
推荐专业知识







- 136氪首发 | 瞄准企业“流 3926
- 2失联37天的私募大佬现身,但 3217
- 3是时候看到全球新商业版图了! 2808
- 436氪首发 | 「微脉」获1 2759
- 5流浪地球是大刘在电力系统上班 2706
- 6招商知识:商业市场前期调研及 2690
- 7Grab真开始做财富管理了 2609
- 8中国离硬科幻电影时代还有多远 2328
- 9创投周报 Vol.24 | 2183
- 10微医集团近日完成新一轮股权质 2180



